Quelques Tutoriels à votre disposition
Initiation à l’Utilisation du Cluster Muse
Connexion au calculateur haute performance Muse
La connexion au cluster de calcul haute performance se fait via le protocole SSH. Le nom d’hôte de la machine de connexion est «muse-login.meso.umontpellier.fr».
Suivant votre système d’exploitation, vous pouvez vous y connecter comme suit :
Sous linux :
Ouvrir une connexion ssh dans un terminal en tapant la commande suivante : « ssh -X « nom_utilisateur »@muse-login.meso.umontpellier.fr »
(L’option -X permet d’avoir un affichage graphique)
Sous windows :
Installer le logiciel putty , ouvrir putty en remplissant les champs de configuration comme suit :
– Hostname : « muse-login.meso.umontpellier.fr »
– Connection type : « SSH »
Cliquer ensuite sur « Open » et remplir les champs « login as» et « password » avec ceux fournis lors de la création du compte.
Vous êtes connecté au cluster Muse.
Si vous souhaitez obtenir un affichage graphique, installer le logiciel Xming et l’ouvrir. Rajouter l’option suivante avant d’appuyer sur « Open » dans putty :
« Category > Connection > SSH > X11 > Cocher Enable X11 forwarding »
Sous Mac :
Installer Xquartz et l’ouvrir
Entrer la commande :
ssh -X « nom_utilisateur »@muse-login.meso.umontpellier.fr
Quel que soit votre système d’exploitation, lors de la première connexion pensez à utiliser la commande « passwd » sur le cluster pour changer le mot de passe par défaut.
Présentation du cluster Muse & bonnes pratiques
Nous organisons régulièrement des sessions de formation pour « présenter le calculateur » et « comment lancer son premier job ».
Les supports de présentation sont disponibles, n’hésitez pas à les utiliser.
Rappel des points de vigilance :
–> Les machines muse-login (muse-login01 et muse-login02) sont les interfaces utilisateur de connexion au cluster.
Les jobs sont lancés depuis muse-login mais ne doivent pas être exécutés sur muse-login, mais via le gestionnaire de job SLURM.
Une charge trop importante des serveurs frontaux pénalise l’ensemble des utilisateurs, ce qui nous amène à supprimer ces jobs. plus d’information sur l’utilisation du gestionnaire de tâche SLURM plus bas.
–> espace de stockage: Il y a plusieurs espaces de stockage (voir détail ci-dessous). Les fichiers déposés sur le répertoire « scratch » sont temporaires pour effectuer vos calcul, et sont automatiquement supprimés à 60 jours. Les documents destinés à être conservés doivent être déposés sur votre répertoire « home ».
Premier pas sous un environnement Linux
Le cluster est sous Linux et ne possède pas d’interface graphique. A cet effet, voici une liste de commandes vous permettant de réaliser des actions basiques une fois connecté :
– ls : liste le contenu de votre répertoire
– cd « chemin_du_dossier » : pour se déplacer dans les sous dossiers de votre répertoire
– pwd : affiche le répertoire courant
– mv (ou cp) fichier_base fichier_destination : déplace (ou copie) un fichier
– mkdir « nom_dossier » : crée un sous-dossier dans le répertoire courant
– vim (ou nano) « fichier_texte » : édite un fichier (le crée si il n’existe pas)
– cat « fichier_texte » : affiche le contenu du fichier
N’hésitez pas à consulter le site suivant pour découvrir d’autres possibilités https://doc.ubuntu-fr.org/tutoriel/console_commandes_de_base
Le gestionnaire d’environnement module
Le gestionnaire d’environnement module permet de configurer les variables d’environnements adaptées à votre programme. Les modules fonctionnent par groupe. Pour obtenir l’ensemble des modules d’un groupe, il vous faut utiliser la commande « module load group ». Par exemple, pour le module le plus courant est « module load cv-standard » et ensuite vous pouvez taper « module available » pour obtenir la liste des modules présents dans le groupe « cv-standard ». Sélectionnez ensuite le compilateur, la bibliothèque MPI et les bibliothèques de routines scientifiques selon vos besoins et chargez-les :
GNU compilers, Intel icc/icpc/ifort
OpenMPI, Intel MPI, mpich, mvapich, mvapich2, etc…
Blas, Lapack, hdf5, fftw3, etc…
Exemple :
$ module load openmpi/2.0.1
$ module load gcc/4.9.3
Les principales options sont :
$ module available : Liste des modules disponibles.
$ module list : Liste des modules chargés dans votre environnement.
$ module show : Description du module.
$ module purge : Supprime tous les modules de votre environnement.
$ module add/load : Charge le module dans votre environnement.
$ module rm/unload : Supprime le module de votre environnement.
La liste des modules installés est disponible en bas de page
Utilisation du gestionnaire de tâche SLURM sur le cluster MUSE
Il y a deux modes d’exécution d’un calcul :
⇒ Un mode d’exécution en temps réel avec la commande srun
⇒ Un mode d’exécution différé avec la commande sbatch.
Avec la commande « srun » les paramètres d’exécution sont renseignés en ligne. Avec la commande « sbatch » les paramètres d’exécution sont renseignés dans un fichier batch.
Nous vous recommandons d’utiliser le mode différé.
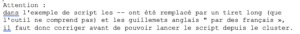
Exemple de fichier batch :
#!/bin/sh
#SBATCH –job-name=test
#SBATCH -N 3
#SBATCH -n 9
#SBATCH –ntasks-per-node=3
#SBATCH –ntasks-per-core=1
#SBATCH –partition=defq
#SBATCH –time=01:00:00echo « Running on: $SLURM_NODELIST »
mpirun ./mon_programme.exe
Le job nommé ‘test’ va s’exécuter sur 3 cœurs * 3 nœuds soit 9 cœurs au total. La queue de calcul est ‘defq’ et avec une durée limite d’une heure.
La variable d’environnement $SLURM_NODELIST permet de récupérer le nom des nœuds sur les quels le job est exécuté.
Chaque ligne commençant par « #SBATCH » décrit un paramètre SLURM. Il est important que ces paramètres se trouvent en haut de fichier avant la première instruction à exécuter.
Remarque : Pour certains paramètres (mais pas tous) il existe deux écritures différentes. Une écriture longue et une écriture courte. Ainsi le nom du job peut être déclaré par ‘–job-name=test’ ou par ‘-J test’. Le nombre de nœuds de calcul peut être déclaré par ‘–nodes=3’ ou par ‘-N 3’ (notez que la syntaxe est un peu différence selon le format utilisé).
Attention au temps de calcul par défaut les queues de calcul « defq » et « thau-smp » ont un temps d’allocation de 7 jours par défaut.
Avant de lancer votre Job, le système vérifie que vous disposez du quota suivant : (7 jours * 24heures * nombres de coeurs demandés pour votre job)
Si vous ne disposez pas du quota nécessaire vous aurez les messages d’erreur suivant : « queued and waiting for resources » (avec la commande squeue : « QOSGrpCPUMinutesLimit »)
Pour pallier à cela vous devez ajouter la variable « –time=HH:MM:SS » en indiquant le nombre d’heures que vous estimez nécessaire à la réalisation du calcul
Notre conseil : arrondir au-dessus. Il ne s’agit que d’une estimation et non pas un décompte d’heure réel
Seules les heures réellement calculées seront déduites de votre quota
Le tableau ci-dessous récapitule les principaux paramètres :
| Format long | Format court | Description |
|---|---|---|
| --job-name | -J | Nom du job |
| --account | Nom du projet | |
| --nodes | -N | Nombre de nœuds |
| --ntasks | -n | Nombre de tâches au total |
| --ntasks-per-node | Nombre de tâches par nœud (doit correspondre au nombre total de tâches divisé par le nombre de nœuds) | |
| --ntasks-per-core | Nombre de tâches par cœurs (en général 1) | |
| --partition | -p | Groupe de machines sur lequel le job va tourner |
| --mem | Mémoire réservée par nœud en MegaBytes? (Si cette option n’est pas spécifiée la totalité de la mémoire du nœud est allouée) | |
| --time | -t | Le temps maximum durant lequel le job va tourner (Passé ce délai, le job est automatiquement arrété) |
| --output | -o | Le nom du fichier oû sera écrit les sorties. Par défaut slurm-%j.out ou '%j' est le numéro du job |
| --error | -e | Le nom du fichier oû seront écrit les messages d'erreur. Par défaut slurm-%j.err ou '%j' est le numero du job |
| --time=HH:MM:SS | -t HH:MM:SS | Temps limite de votre job en heures, minutes et secondes. |
| --mail-user | Adresse mail à laquelle les notifications seront envoyées |
Si vous exécutez un job OpenMP renseignez la variable d’environnement OMP_NUM_THREADS dans votre fichier batch à la suite des paramètres.
Exemple : export OMP_NUM_THREADS=12
Remarque : La commande « srun » peut être utilisée à l’intérieur d’un fichier batch que l’on exécute avec la commande « sbatch ». Dans ce cas les paramètres peuvent être renseignés à la suite de la commande srun.
Autres commandes SLURM utils :
$ squeue : Affiche la liste des jobs avec leurs états
$ scancel 221 : Supprime le job 221 de la queue
$ sinfo : Liste les partitions de calcul avec leurs caractéristiques
$ scontrol show jobid 221 : Affiche le détail du job 221
Quelques ressources d’initiation à l’utilisation du Cluster
1-Presentation_cluster_Muse
1-TP-Environment_module
1-TP-SLURM
Décompte des heures sur le Cluster MUSE
La commande « mon_nbre_heures » est disponible à tout moment sur « muse-login » pour vous permettre de vérifier votre consommation d’heure de calcul.
Exemple de scripts optimisés pour le cluster MUSE
Dans cette archive vous trouverez :
– un exemple de job MPI
– un exemple de job OpenMP
– un exemple de job MPI + OpenMP
A noter : Les paramètres utilisés dans les 3 exemples, optimisés pour l’utilisation de slurm, sont transférable à l’ensemble de vos job sous Slurm.
Comment organiser ses fichiers
| Montage | Description | Utilisation | Quota | Péremption |
|---|---|---|---|---|
| /home | NFS | Fichiers de configuration globale code, bibliothèques | Partagé avec le groupe (en général 50GigaOctet partagé) | Non |
| /nfs/work | NFS | Fichiers de configuration des travaux Données d'entrée et de sortie Copie des exécutables | Partagé avec le groupe (en général 50GigaOctet partagé) | Non |
| /lustre/scratch | lustre | Exécution des travaux Donnée temporaires Le répertoire Scratch est à utiliser impérativement pour l'exécution des jobs. Les résultats peuvent ensuite être déplacés vers les répertoires /home ou /work | Non | 2 mois Au delà, les données sont effacées |
Il faut TOUJOURS utiliser votre répertoire de scratch pour l’exécution de vos jobs et ensuite déplacer les résultats que vous souhaitez conserver sur votre /home ou /work.
Utilisation des noeuds GPU de Visualisation
Voici une documentation pour l’utilisation des noeuds de Visualisation.
L’utilisation des nœuds de visu nécessite le lancement du script suivant : Visu-slurm
Environnement logiciel du Cluster Muse
Les logiciels sont installés sous forme de modules dans le répertoire commun. La liste suivante (mise à jour le 19/05/2020) n’est qu’indicative, car nous effectuons des mise à jour quotidiennement :
| - Modules arborescence local : Cast3M/2019 GMT/5.2.1 ImageMagick/7.0.3-10 JDK/jdk.8_x64 Nlopt/2.6.1 R/3.6.1 R/3.6.1-tcltk R/4.0.2 VTK/7.1.1 VTK/8.1.1 automake/1.16.1 flex/2.6.4 gdal/2.1.2 gdal/3.0.1 geos/3.7.2 ghmm/1.0 go/12.9 gromacs/2019.4 gsl/2.3 gsl/2.4 intel/opencl-1.2-6.4.0.37 jre/jre.8_x64 julia/1.1.0 knem/1.1.2 lapack-with-PIC/3.7.0 libarchive/3.3.2 netcdf/4.4.1.1 pi4u/torc proj/6.1.1 python/3.5.2 python/3.5.2-bz2 python/3.7.2 python/3.8.2 python/3.8.3_shared_library python/Anaconda/3-5.1.0 sage/7.6 scite/3.7.1 singularity/2.2.1 singularity/2.4 singularity/2.6 singularity/3.3 singularity/3.5 squashfs/4.3 swig/3.0.11 vmd/1.9.3 xmgrace/5.1.25 - Modules arborescence cv-standard : R/3.3.1 R/3.4.3 blas/3.6.0 boost/1.61.0(default) boost/icc16/1.61.0 boost/impi/icc16/1.61.0 boost/mvapich2/1.61.0 boost/mvapich2/icc16/1.61.0 boost/openmpi/1.61.0 boost/openmpi/icc16/1.61.0 cmake/3.6.0(default) fftw2/2.1.5(default) fftw3/3.3.5(default) fftw3/3.3.8-test fftw3/mvapich2/3.3.5 fftw3/openmpi/3.3.5 fftw3-mvapich2/mvapich2/3.3.5 fftw3-openmpi/openmpi/3.3.5 gcc/4.9.3(default) gcc/5.3.0 gcc/6.1.0 gcc/7.5.0 gdb/7.11 git/2.9.3 hdf5/1.10.1(default) hdf5/1.8.17 hdf5/gcc49/1.10.1 hdf5/gcc49/1.8.17(default) hdf5/gcc53/1.10.1 hdf5/gcc53/1.8.17 hdf5/gcc61/1.10.1 hdf5/gcc61/1.8.17 hdf5/icc16/1.10.1 hdf5/icc16/1.8.17 hdf5/impi/icc16/1.10.1 hdf5/impi/icc16/1.8.17 hdf5/mvapich2/1.10.1 hdf5/mvapich2/1.8.17 hdf5/mvapich2/gcc49/1.10.1 hdf5/mvapich2/gcc49/1.8.17 hdf5/mvapich2/gcc53/1.10.1 hdf5/mvapich2/gcc53/1.8.17 hdf5/mvapich2/gcc61/1.10.1 hdf5/mvapich2/gcc61/1.8.17 hdf5/mvapich2/icc16/1.10.1 hdf5/mvapich2/icc16/1.8.17 hdf5/openmpi/1.10.1 hdf5/openmpi/1.8.17 hdf5/openmpi/gcc49/1.10.1 hdf5/openmpi/gcc49/1.8.17 hdf5/openmpi/gcc53/1.10.1 hdf5/openmpi/gcc53/1.8.17 hdf5/openmpi/gcc61/1.10.1 hdf5/openmpi/gcc61/1.8.17 hdf5/openmpi/icc16/1.10.1 hdf5/openmpi/icc16/1.8.17 hwloc/1.11.2 intel/advisor/32/2016.3.210 intel/advisor/32/2017.1.132 intel/advisor/64/2016.3.210 intel/advisor/64/2017.1.132 intel/clck/64/2016.3.210 intel/clck/64/2017.1.132 intel/clck/mic/2016.3.210 intel/clck/mic/2017.1.132 intel/compiler/32/2016.3.210 intel/compiler/32/2017.1.132 intel/compiler/64/2016.3.210 intel/compiler/64/2017.1.132 intel/daal/32/2016.3.210 intel/daal/32/2017.1.132 intel/daal/64/2016.3.210 intel/daal/64/2017.1.132 intel/inspector/32/2016.3.210 intel/inspector/32/2017.1.132 intel/inspector/64/2016.3.210 intel/inspector/64/2017.1.132 intel/ipp/32/2016.3.210 intel/ipp/32/2017.1.132 intel/ipp/64/2016.3.210 intel/ipp/64/2017.1.132 intel/itac/64/2016.3.210 intel/itac/64/2017.1.132 intel/mkl/32/2016.3.210 intel/mkl/32/2017.1.132 intel/mkl/64/2016.3.210 intel/mkl/64/2017.1.132 intel/mkl/mic/2016.3.210 intel/mkl/mic/2017.1.132 intel/mpi/64/2016.3.210 intel/mpi/64/2017.1.132 intel/omnipath/64/libpsm2-10.3.8-3 intel/runtime/32/2016.3.210 intel/runtime/32/2017.1.132 intel/runtime/64/2016.3.210 intel/runtime/64/2017.1.132 intel/tbb/32/2016.3.210 intel/tbb/32/2017.1.132 intel/tbb/64/2016.3.210 intel/tbb/64/2017.1.132 intel/vtune/32/2016.3.210 intel/vtune/32/2017.1.132 intel/vtune/64/2016.3.210 intel/vtune/64/2017.1.132 lapack/3.6.1 matplotlib/py27/1.5.3 mvapich2/gcc49/2.2rc2 mvapich2/gcc53/2.2rc2 mvapich2/gcc61/2.2rc2 mvapich2/psm/2.2rc2 mvapich2/psm/icc16/2.2rc2 mvapich2/psm2/2.2rc2 mvapich2/psm2/gcc49/2.2rc2(default) mvapich2/psm2/gcc53/2.2rc2 mvapich2/psm2/gcc61/2.2rc2 mvapich2/psm2/icc16/2.2rc2 numpy/py27/1.11.2 openblas/0.2.18(default) openmpi/icc17/2.0.1 openmpi/psm2/2.0.1 openmpi/psm2/3.1.6 openmpi/psm2/gcc49/2.0.1(default) openmpi/psm2/gcc49/3.1.6 openmpi/psm2/gcc53/2.0.1 openmpi/psm2/gcc53/3.1.6 openmpi/psm2/gcc61/2.0.1 openmpi/psm2/gcc61/3.1.6 openmpi/psm2/gcc75/3.1.6 openmpi/psm2/icc17/2.0.1 openmpi/psm2/icc17/3.1.6 openmpi/psm2/old.version python/2.7.12(default) qt/gcc/4.8.6 scalapack/mvapich2/2.0.2 scalapack/openmpi/2.0.2 scilab/5.5.2 scipy/py27/0.18.1 valgrind/3.11.0 - Modules arborescence cv-advanced : M2/1.9.2 gromacs/openmpi-2.0.1-psm2/gcc-6.1.0/5.1.4 metis/4.0.3(default) metis/5.1.0 metis/icc16/4.0.3 metis/icc16/5.1.0 namd/impi-5.1.3/gcc-6.1.0/2.11 namd/ompi-2.0.1-psm2/2.11 paraview/gcc49-ompi-qt-mesa/5.1.0 parmetis/impi/icc16/4.0.3 parmetis/mvapich2/4.0.3 parmetis/mvapich2/icc16/4.0.3 parmetis/openmpi/4.0.3 parmetis/openmpi/icc16/4.0.3 scotch/6.0.4(default) scotch/icc16/6.0.4 |
Vous pouvez installer des logiciels dans votre espace utilisateur.
Cas des logiciels payants : l’utilisateur doit s’acquitter des droits de licence.
Possibilité d’utiliser des logiciels avec serveur de licence externe (Exemple : MatLab)
Copier des fichiers de façon sécurisée depuis le Cluster Muse
Ce fichier vous permet de copier des données depuis le cluster Muse vers votre machine LINUX.
Il vous faut modifier les champs USER, DOSSIER_CLUSTER et DOSSIER_PERSO et ensuite le lancer avec la commande « bash rsync ».
Il est vivement conseillé d’utiliser ce script lors de téléchargement de fichiers volumineux.